Today marks a major update release for HybPiper, version 1.3, which I am calling The Herbarium Update. Many of the features added and bugs fixed have to do with processing targeted sequencing data that came from herbarium specimens.
Herbarium material starts with degraded DNA, and frequently has fragment sizes less than 100 bp. When HybSeq libraries are prepared from this material and sequenced on a platform with > 100 bp reads, the reads themselves will be truncated. The lower depth will also result in shorter contigs assembled by SPAdes within HybPiper.
The biggest issue dealing with these short contigs is when they only have a portion of one exon in a multi-exon gene. With the intron/exon boundary missing, Exonerate was proposing multiple (sometimes overlapping) alignments. As a result, HybPiper was generating incorrect coding sequence, sometimes with contigs repeated or in the wrong order. The fix was to select only one contig for each region, ignoring other possible alignments. This can result in shorter sequence recovery, but the region that is recovered will at least be correct!
Sequences recovered from fresh tissue (which normally have long contigs and sufficient depth) should not be affected by this issue.
In addition, I added a new “heatmap” script thanks to suggestions made by Paul Wolf. The new script uses ggplot and is a lot simpler to use and customize.
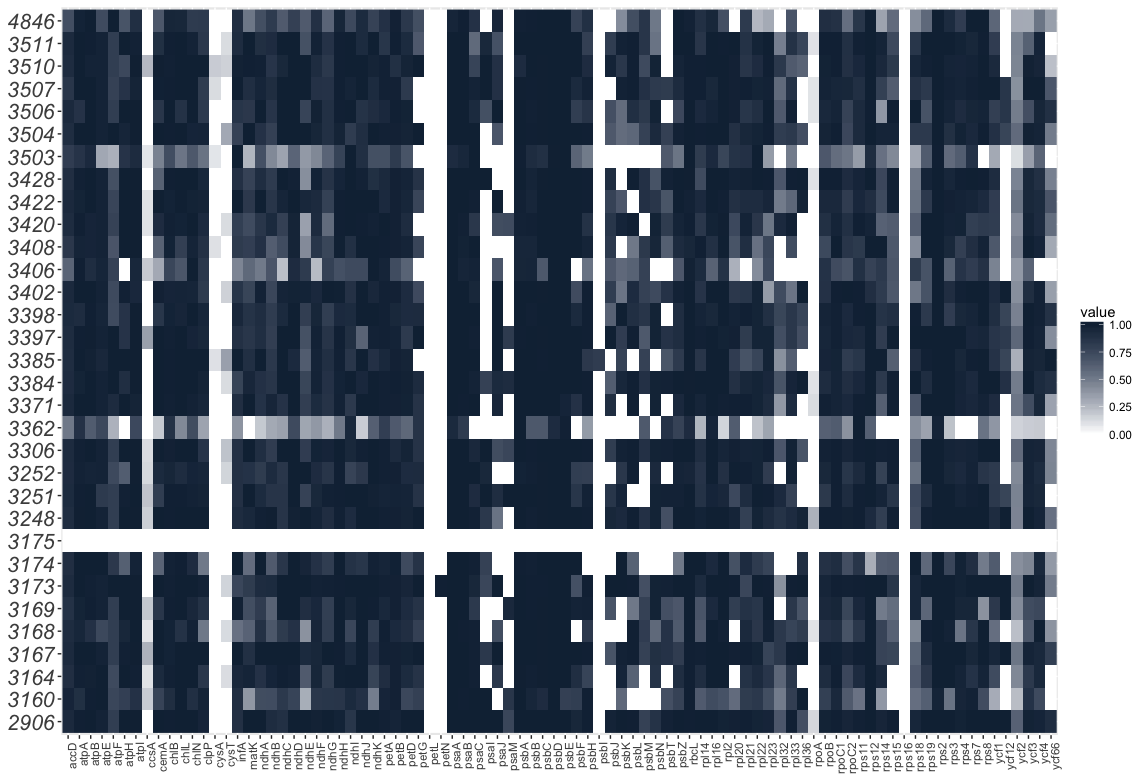
I am keeping the old script for now, because although the ggplot version looks great as a PNG, there are issues exporting to a “vectorized” PDF. The old script still works for this, so I am keeping it.
The new version of HybPiper is available on GitHub. I’m happy to see that HybPiper is getting some use in published papers!